influenza NP 1918 descended¶
An analysis of the evolution of nucleoprotein (NP) from influenza viruses (human and swine) descended from the 1918 virus.
This is the analysis used to create the mutational paths in Gong and Bloom, 2014.
This analysis is found in the ./examples/influenza_NP_1918_Descended/ subdirectory of the main mutpath repository on GitHub.
This analysis was performed by Jesse Bloom.
Software used¶
- mutpath (version 0.1): the main software package for building the trajectory from the BEAST output.
- Python (version 2.6.8)
- BEAST (version 1.8pre Prelease r5356)
- BEAGLE (revision 1093)
- RAxML (version 7.6.3)
- TreeAnnotator (version 1.7.5)
- FigTree (version 1.4.0): to visualize phylogenetic trees
- GraphViz (version 2.30.1): to visualize mutational trajectories
- mapmuts (version 1.0): used by the Python scripts to parse the sequence sets.
Construction of sequence sets¶
The first step is construction of aligned coding sequence sets for the human and swine lineages. The ultimate set of aligned sequences are in Combined_NP_nts.fasta and Combined_NP_proteins.fasta.
Separate analyses were done for the human and swine lineages. The sets are constructed by the scripts get_human_seqs.py and get_swine_seqs.py which construct the files Human_NP_nts.fasta, Human_NP_proteins.fasta, Swine_NP_nts.fasta, and Swine_NP_proteins.fasta. The swine and human files were then concatenated to create Combined_NP_nts.fasta and Combined_NP_proteins.fasta. These scripts use the following input files:
- NPseqs.fasta is the set of all unique full-length influenza A coding DNA sequences as downloaded from the Influenza Virus Resource on June-25-2013.
- Aichi68-NP.fasta is the coding DNA sequence for A/Aichi/2/1968 (H3N2) NP as taken from reverse-genetics plasmid pHWAichi68-NP.
- JVI_82_8947_Anomalies.txt is a list of the strain names for the sequences identified as anomalous (either frozen in time or recombinant) in Appendices 1 and 2 of Krasnitz et al, 2008.
- JDB_Anomalies.txt is a list of strain names that I (Jesse D. Bloom, JDB) have found appear to be anomalous based on their tree positioning using analyses with RAxML and Path-O-Gen as described below.
Human lineages¶
The retained sequences are all human NPs that are descended directly from the 1918 viruses in the human lineage. These are:
- H1N1 from 1918 to 1957: these include the 1918 virus and its direct descendents.
- H2N2 from 1957 to 1968: the “Asian flu” of 1957 involved replacement of several genes from human H1N1, but the NP in this H2N2 was derived from the previous human H1N1.
- H3N2 from 1968 to 2013: the “Hong Kong flu” of 1968 involved replacement of several genes from human H2N2, but the NP in this H3N2 was derived from the previous human H2N2.
- H1N1 from 1977 to 2008: the seasonal H1N1 reappeared in 1977, apparently due to revival of the human H1N1 from about 1954. The dates for these sequences therefore need to have 24 years substracted in order to conform to a molecular clock (see dos Reis et al, 2009). This seasonal H1N1 went extinct in early 2009 due to the appareance of the 2009 swine-origin pandemic H1N1.
Only sequences encoding unique proteins are retained, and a maximum of 5 sequences per year of any given subtype are retained to make the trees not too large. Anomalous sequences are removed, based on those specified by Krasnitz et al, 2008, and also those that I find to be in strong violation of the molecular clock based on an analysis of non-date-stamped trees built with RAxML with Path-O-Gen. Specifically, the analysis was done as follows:
Extract the human NP sequences descended from the 1918 virus into Human_NP_nts.fasta and Human_NP_proteins.fasta:
python get_human_seqs.pyBuild a RAxML tree (no date stamping) to allow visual inspection for possible outliers, and write this tree to the RAxML_output subdirectory:
/Users/jbloom/standard-RAxML-master/raxmlHPC -w /Users/jbloom/mutpath/examples/influenza_NP_1918_Descended/RAxML_output -n Human_NP_nts -p 1 -m GTRCAT -s Human_NP_nts.fasta /Users/jbloom/standard-RAxML-master/raxmlHPC -w /Users/jbloom/mutpath/examples/influenza_NP_1918_Descended/RAxML_output -n Human_NP_proteins -p 1 -m PROTCATJTT -s Human_NP_proteins.fastaThe trees were then visually inspected using Path-O-Gen, and clear outliers from the molecular clock were removed by adding them to JDB_Anomalies.txt and re-running the analysis.
Swine lineages¶
The retained sequences are swine NPs that are descended directly from the 1918. These are taken only from North American viruses (USA, Canada; viruses from Mexico not included since many seem anomalous). According to Brockwell-Staats et al, 2009, the North American viruses are predominantly the classical H1N1 lineage from 1918 to 1998, and from then on the NP is maintained even as some of them reassort other viral genes.
Only sequences that encode unique proteins are retained. A maximum of 5 sequences per year of any given subtype are retained to make the trees not too large. Anomalous sequences are removed, based on those specified by Krasnitz et al, 2008, and also those that I find to be in strong violation of the molecular clock based on an analysis of non-date-stamped trees built with RAxML with Path-O-Gen. Also, there are some viruses that are not descended from the 1918 among the swine North American viruses – they are probably mixed with other lineages or come from avian – these are also removed. Specifically, the analysis was done as follows:
Extract the swine NP sequences descended from the 1918 virus into Swine_NP_nts.fasta and Swine_NP_proteins.fasta:
python get_swine_seqs.pyBuild a RAxML tree (no date stamping) to allow visual inspection for possible outliers, and write this tree to the RAxML_output subdirectory:
/Users/jbloom/standard-RAxML-master/raxmlHPC -w /Users/jbloom/mutpath/examples/influenza_NP_1918_Descended/RAxML_output -n Swine_NP_nts -p 1 -m GTRCAT -s Swine_NP_nts.fasta /Users/jbloom/standard-RAxML-master/raxmlHPC -w /Users/jbloom/mutpath/examples/influenza_NP_1918_Descended/RAxML_output -n Swine_NP_proteins -p 1 -m PROTCATJTT -s Swine_NP_proteins.fastaThe trees were then visually inspected using Path-O-Gen, and clear outliers from the molecular clock were removed by adding them to JDB_Anomalies.txt and re-running the analysis.
Combined human and swine lineages¶
The human and swine files were combined into Combined_NP_nts.fasta and Combined_NP_proteins.fasta:
cat Human_NP_nts.fasta Swine_NP_nts.fasta > Combined_NP_nts.fasta
cat Human_NP_proteins.fasta Swine_NP_proteins.fasta > Combined_NP_proteins.fasta
RAxML was then used to build a tree (no date stamping) with:
/Users/jbloom/standard-RAxML-master/raxmlHPC -w /Users/jbloom/mutpath/examples/influenza_NP_1918_Descended/RAxML_output -n Combined_NP_nts -p 1 -m GTRCAT -s Combined_NP_nts.fasta
/Users/jbloom/standard-RAxML-master/raxmlHPC -w /Users/jbloom/mutpath/examples/influenza_NP_1918_Descended/RAxML_output -n Combined_NP_proteins -p 1 -m PROTCATJTT -s Combined_NP_proteins.fasta
This tree was then visually analyzed using Path-O-Gen to confirm that it appears to be fairly clock-like given the date stamps for the tips.
Mapping the mutational trajectory¶
Running BEAST¶
The file Combined_NP_proteins.xml was constructed from the sequences in Combined_NP_proteins.fasta as a BEAST input file using a combination of BEAUTI and hand-annotation. This XML file specifies date-stamped sequences, a strict molecular clock, and a JTT model of substitution.
This file was then used as the input for four different runs of BEAST (version 1.8pre Prelease r5356) using the BEAGLE (revision 1093) library, which were performed in the subdirectories run1/, run2/, etc. These runs were performed on the FHCRC’s rhino cluster using sbatch with the command:
sbatch run.sbatch
where the contents of the run.sbatch file was as follows:
#!/bin/sh
#SBATCH
#PBS -l walltime=480:00:00
echo "Starting..."
java -Xmx4048m -Xms4048m -Djava.library.path=/home/jbloom/BEAGLE_libs/lib -cp ~/BEAST/build/dist/beast.jar dr.app.beast.BeastMain -beagle Combined_NP_proteins.xml
echo "Finished."
The identical command was executed in all four run directories.
Inspection of the .log files with Tracer indicated that the runs (each of 20 million steps with trees saved every 10,000 steps) appeared to have equilibrated after about 2.5 million steps (the first 250 saved trees). If these are removed as burn-in and the four runs are combined, the effective sample sizes seem adequate to suggest MCMC convergence.
Each of the .trees files were compacted:
mutpath_compact_trees.py run1/Combined_NP_proteins.trees
mutpath_compact_trees.py run2/Combined_NP_proteins.trees
mutpath_compact_trees.py run3/Combined_NP_proteins.trees
mutpath_compact_trees.py run4/Combined_NP_proteins.trees
This created the files run1/Combined_NP_proteins_compact.trees, etc.
Note that these .trees files are not included in the mutpath repository on GitHub due to large file sizes.
Building the trajectories¶
Two trajectories were then built:
- For human H3N2, the trajectory from A/Aichi/2/1968 (H3N2) to A/Texas/JMM_49/2012 (H3N2).
- For swine, the trajectory from A/swine/Wisconsin/1/1957 (H1N1) to A/swine/Indiana/A00968365/2012 (H1N1).
The human H3N2 trajectory was built using the command:
mutpath_get_paths.py get_paths_infile_human_H3N2.txt
mutpath_make_digraph.py make_digraph_infile_human_H3N2.txt
where the contents of get_paths_infile_human_H3N2.txt are:
# input file to mutpath_get_paths.py
intreefiles run1/Combined_NP_proteins_compact.trees run2/Combined_NP_proteins_compact.trees run3/Combined_NP_proteins_compact.trees run4/Combined_NP_proteins_compact.trees
burnin 250
mergedtreesfile merged_Combined_NP_proteins_compact.trees
fastafile Combined_NP_proteins.fasta
seqtype protein
startseq 1968.50_COUNT1_STRAIN_A/Aichi/2/1968_HOST_Human_SUBTYPE_H3N2_DATE_1968.50
endseq 2012.93_COUNT98_STRAIN_A/Texas/JMM_49/2012_HOST_Human_SUBTYPE_H3N2_DATE_2012.93
mutpathsfile human_H3N2_mutpaths.txt
and the contents of make_digraph_infile_human_H3N2.txt are:
# input file to mutpath_make_digraph.py
mutpathfile human_H3N2_mutpaths.txt
translateseqs False
dotfile human_H3N2_trajectory.dot
minweight 0.01
labelcutoff 0.6
nodenamefile None
mutationdates human_H3N2_mutationdates
lasttipdate 2012.93
persistencefile human_H3N2_persistence.txt
The swine trajectory was built using:
mutpath_get_paths.py get_paths_infile_swine.txt
mutpath_make_digraph.py make_digraph_infile_swine.txt
where the input files get_paths_infile_swine.txt and make_digraph_infile_swine.txt are modified to specify the correct swine sequences and dates.
The key output of these runs are the DOT files displaying the trajectories, which can be visualized using GraphViz:
human_H3N2_trajectory.dot
swine_trajectory.dot
These DOT files were opened with GraphViz and used to save PDF and JPG files:
human_H3N2_trajectory.pdf
swine_trajectory.pdf
human_H3N2_trajectory.jpg
swine_trajectory.jpg
These images are shown below.
swine influenza mutational trajectory¶
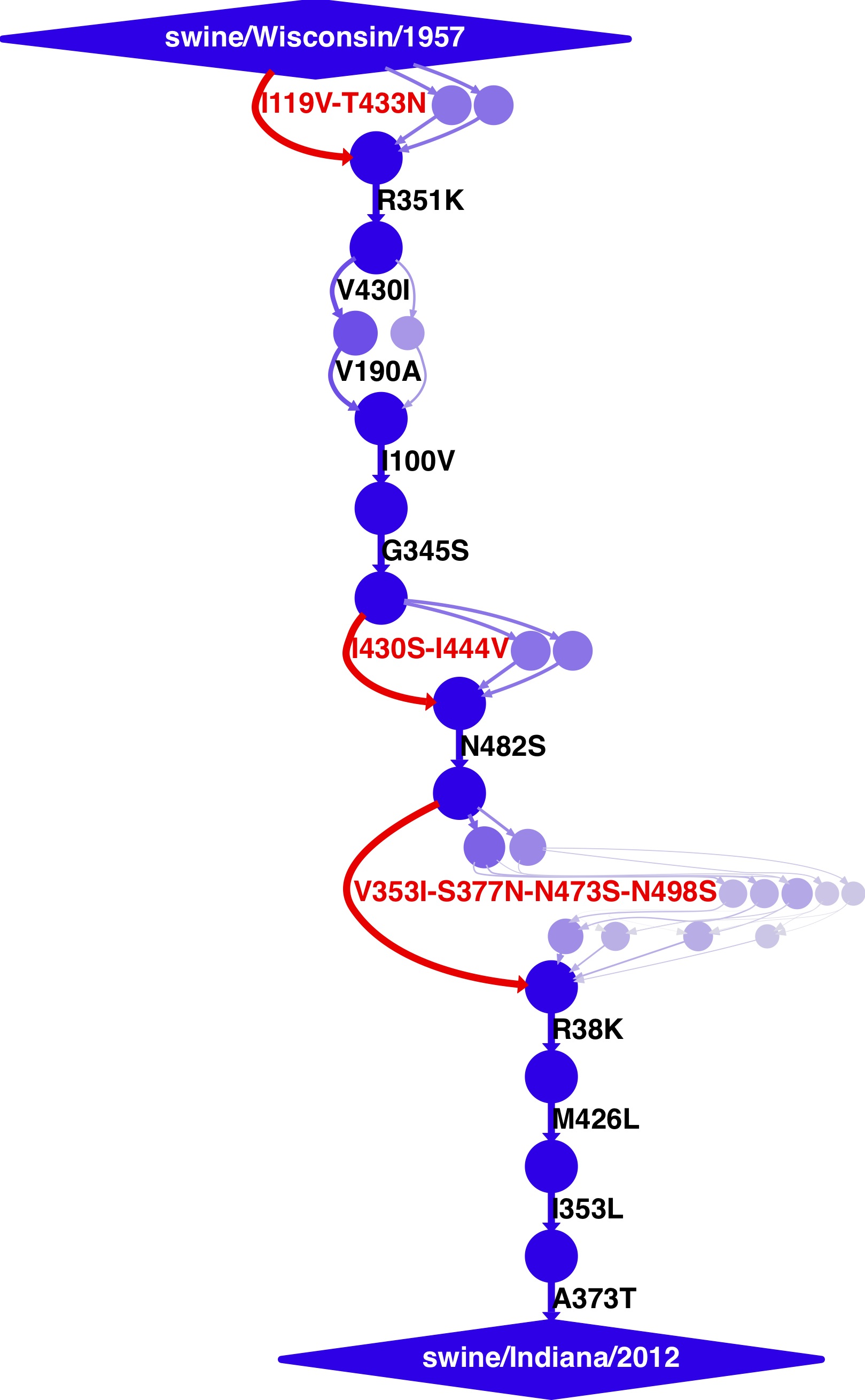
Mutational trajectory for swine influenza.
Human H3N2 mutational trajectory¶
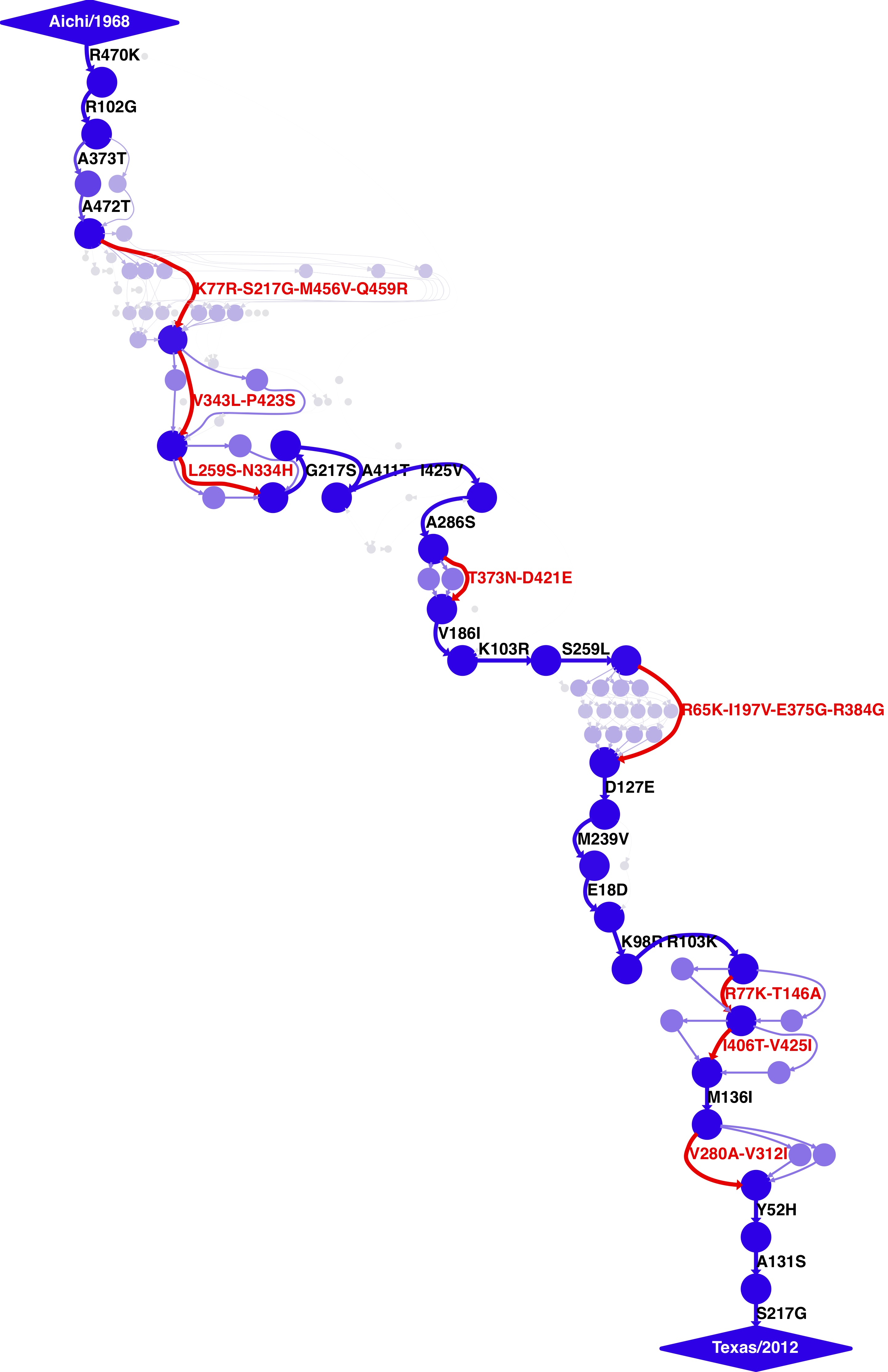
Mutational trajectory for human H3N2. Note that the beginning of this trajectory is slightly different from that in Gong et al, 2013 possibly because of the inclusion of additional sequences from H2N2 that contribute to the early part of the phylogenetic tree.
Building the maximum clade credibility tree¶
In addition, the mutpath_get_paths.py runs created the merged .trees file merged_Combined_NP_proteins.fasta, which was used to build the maximum clade credibility tree maxcladecredibility.trees using TreeAnnotator (version 1.7.5) with the command:
~/BEASTv1.7.5/bin/treeannotator merged_Combined_NP_proteins_compact.trees maxcladecredibility.trees
This tree was then manually edited using mutpath_annotate_tree.py to create annotated_maxcladecredibility.trees by the command:
mutpath_annotate_tree.py annotate_tree_infile.txt
The output file annotated_maxcladecredibility.trees was then opened in FigTree where it was saved to handannotated_maxcladecredibility.trees and annotated further by hand. The image was then saved using FigTree as handannotated_maxcladecredibility.pdf and then converted to a JPG with:
convert -density 400 handannotated_maxcladecredibility.pdf handannotated_maxcladecredibility.jpg
This tree is shown below:
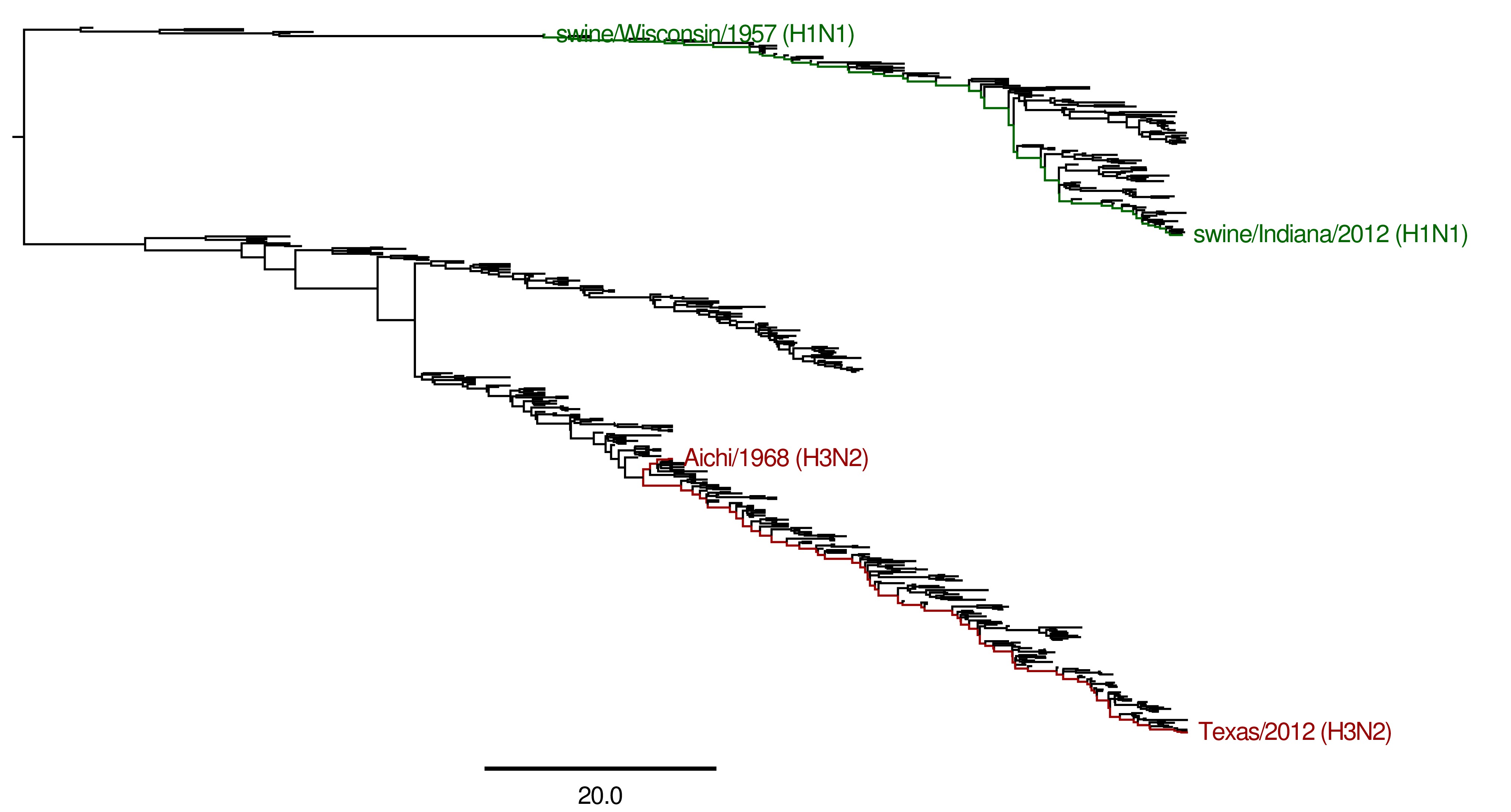
Maximum clade credibility tree of NPs descended from 1918 virus. The swine trajectory is in green and the human H3N2 in red.